7. ВЛИЯНИЕ СУШКИ И ТЕРМИЧЕСКОГО РАЗЛОЖЕНИЯ ТОПЛИВА НА ПРОЦЕССЫ ГОРЕНИЯ И ГАЗИФИКАЦИИ
Мы уже отмечали, что рациональная организация процесса горения и газификации подавляющего большинства твердых топлив в слое сводится к организации горения и газификации твердого коксового остатка, основой которого является углерод*. Прямым непосредственным доказательством этого положения, например, в части интенсивности процесса горения слоя топлива в диффузионной области является тот факт, что в случае правильной постановки исследования размер кислородной зоны при равных размерах частиц практически одинаков для электродного угля, графита, древесного угля, торфяного кокса и др. аналогичных топлив, лишенных значительной части летучих веществ, и для натуральных углей, сжигаемых в системе с неограниченным воспламенением. Нетрудно показать, что в этих условиях при достаточной высоте слоя топлива в кислородную зону всегда поступает прогретое обуглероженное топливо, лишенное основной массы летучих веществ. При сжигании топлива в условиях ограниченного воспламенения летучие вещества оказывают более существенное влияние, так как в этом случае в кислородную зону поступают все продукты процессов сушки и термического разложения топливной массы, а горение топливных частиц иногда начинается еще до практического завершения указанных процессов. В этом случае резко изменяется тепловой режим процесса, а при переходе его в результате снижения температуры горения в кинетическую или переходную области существенно меняется и скорость процесса и, в некоторой мере, химический механизм взаимодействия топлива с кислородом дутья. В диффузионной области горения кислородная зона существенно сокращается при ограниченном воспламенении вследствие расходования кислорода на взаимодействие с горючими летучими веществами, выделяющимися из топлива.
В случае сжигания крупнокускового топлива при сравнительно небольшой высоте слоя влияние летучих веществ на размер кислородной зоны возможно и при неограниченном воспламенении, так как в связи с медленным прогревом кусков топлива возможна резкая разница температур в центре куска и на его горящей поверхности.
Именно по этой причине на практике встречаются (например в слоевых газогенераторах) мокрые в середине, а с поверхности обгоревшие обуглероженные куски торфа.
При всех практически важных условиях, однако, процесс горения углеродной (коксовой) части топлива остается определяющим как для кислородной, так и особенно для восстановительной зон горения и газификации. Как мы увидим в дальнейшем, при горении газовзвеси топлив, богатых летучими веществами, положение может резко измениться в связи с изменением характера и количества образующихся летучих веществ.
Хорошо известно, что процесс взаимодействия углерода с кислородом резко отличается от взаимодействия кислорода с натуральным топливом, имеющим сложную химическую структуру. В случае взаимодействия с молекулярным топливным комплексом большая часть кислорода вначале присоединяется к твердому топливу, которое, разлагаясь затем при повышении температуры, выделяет этот кислород, главным образом в виде молекул водяного пара и лишь в незначительной степени в виде CO2 и СО. Совершенно очевидно, что при нагревании топлива в присутствии кислорода процессы сушки, выделения летучих веществ, окисления топлива и взаимодействия кислорода с углеродным коксовым остатком будут протекать совместно и роль каждого из них будет определяться кинетическими характеристиками соответствующих химических реакций, а также скоростями процессов тепло- и массообмена в этой реагирующей системе.
Наиболее интересно с теоретической и практической точек зрения изучение влияний процессов сушки и выделения летучих веществ на процессы горения и газификации газовзвеси топлив с относительно высоким содержанием летучих веществ. В этом случае в связи с исключительно большими скоростями прогрева топливных частиц, измеряемыми сотнями тысяч и миллионами градусов в секунду, происходит не только резкое усиление взаимодействия различных процессов, но и существенное изменение характера самих процессов, в частности, процесса термического разложения топлива.
Для того чтобы ясно представить влияние процесса термического разложения на горение газовзвеси топлив и затем оценить возможное воздействие этого процесса на п. г. у., необходимо хотя бы кратко остановиться на многочисленных исследованиях, посвященных механизму процесса термического распада (деструкции), вернее химического реагирования органического вещества топлив при нагревании его без доступа свободного кислорода. Будем условно называть этот сложный процесс, связанный и с реакциями распада и с реакциями синтеза, процессом термического разложения (пиролиза) топлива. Мы рассмотрим лишь отдельные работы, связанные главным образом с разложением твердых топлив.
Твердое топливо представляет собой, как правило, механическую смесь нескольких, различных по составу, структуре и строению, сложных органических соединений, в основе которых лежат базисные углеродно-графитовые структуры.
На рис. 94 приведены некоторые возможные схемы структурных моделей углей, из которых видно, что макромолекулярные комплексы угля (назовем их условно «топливными молекулами») формируются на системах углеродно-графитных «полимеров», связанных между собой по той или иной системе гетероатомными или гетероциклическими группами или кислородными мостиками. Каждая топливная молекула, а они достаточно разнообразны, кроме основной циклической базисной «решетки» имеет также своеобразную разветвленную «шубу» из различных атомов и групп атомов, содержащих главным образом углерод, водорода кислород. Эти «мостики» и «шуба» наряду с базисными решетками угля представлены образно в виде зигзагообразных линий на структурной схеме Касаточкина (рис. 94, в).
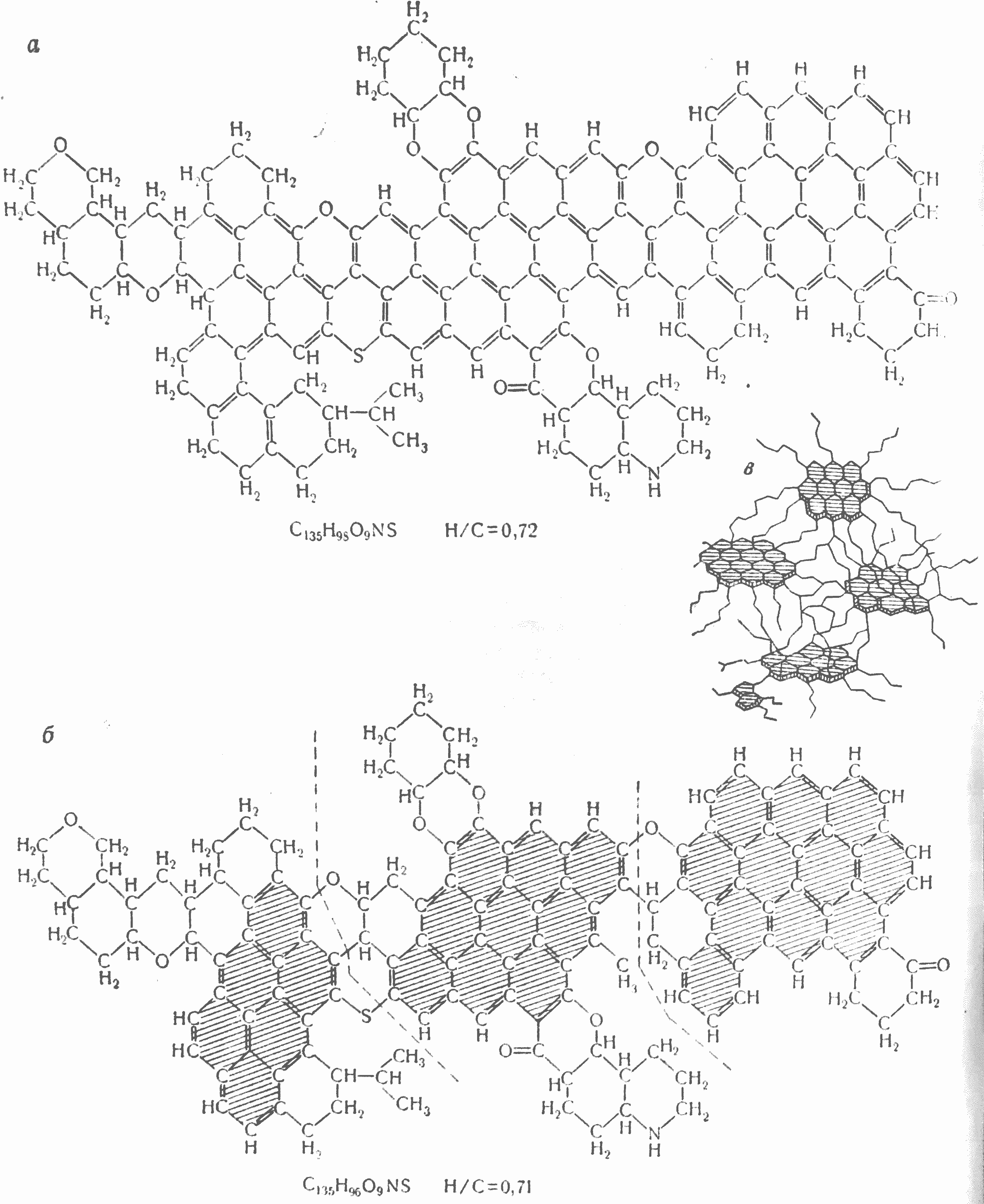
Рис. 94. Структурные модели угольных молекул по Фуксу (а), Кревелену (б) и Касаточкину (в)
Не вдаваясь здесь в детали этого вопроса и анализ различных химических схем Фукса, Кревелена, Касаточкина и др., мы отметим лишь одну важную особенность всех схем, а именно большое количество связей различной энергетической прочности в топливных молекулах. Эта важная особенность служит одновременно и узловым элементом в построении различных химических и кинетических теорий процесса термического разложения твердых топлив. Количество таких теорий в настоящее время достаточно велико, что, конечно, объясняется, в первую очередь, сложностью и самого процесса и строения топлив. Однако, несмотря на большое количество теоретических химических схем разложения топлива, подавляющее большинство их сводится, по существу, к теории
Фукса—Кревелена, которую можно назвать теорией последовательного развития химических реакций разложения — деструкции топлива или, короче, «теорией последовательных реакций». Существо этой теории может быть кратко сформулировано следующим образом: процесс термического разложения с практически достаточной точностью складывается из одной осредненной первичной реакции разложения органического вещества топлива «топливных молекул» и последовательного развития вторичных, третичных и т. д. реакций, протекающих уже с продуктами реагирования топлива. Эта теория химического механизма термического разложения твердых топлив основывается, таким образом, на том, что органическое вещество каждого из механических ингредиентов топлива, т. е. каждая «топливная молекула», может практически разлагаться только с получением вполне определенных первичных продуктов. Такая теоретическая схема процесса исключает возможность изменения качества продуктов и тем самым — возможность управления процессом термического разложения твердых топлив. Эта теория, подкупающая своей простотой и «подтвержденная» многочисленными экспериментальными исследованиями, принята большинством углехимиков различных стран мира.
Кинетическая разработка теории последовательных реакций, естественно, приводит к положению о независимости процесса разложения топлива от скорости его нагревания, так как при наличии одной реакции допускает (как это и делает Крезелен [61]) замену производной по времени в кинетическом уравнении производной по температуре и тем самым исключает влияние времени, а следовательно, и интенсивности нагрева топливной массы на качество и выход продуктов.
Как мы уже отмечали, многочисленные экспериментальные исследования процесса разложения различных твердых топлив хорошо подтверждали тот факт, что в обычных условиях скорость нагрева мало влияет на качество продуктов разложения. Максимальный выход летучих веществ экспериментально получали обычно при медленном нагреве и быстром выводе продуктов из реакционного объема, что также, как будто, подтверждало теорию последовательных реакций, поскольку исключение вторичных реакций в газопаровой фазе приводило к минимальному выходу кокса, а скорость нагрева не влияла на процесс.
Среди большинства углехимиков также широко распространено представление о том, что так называемые битумы (растворимая в органических растворителях часть твердых топлив) являются механической составной частью топлив и удаляются при нагревании топлив в результате испарения и возгонки. Такое представление о битумах, конечно, еще больше сужает возможность регулировки качества и выхода летучих веществ, так как битумы, дающие обычно наибольшее количество смол, должны всегда превращаться в летучие вещества определенного состава при нагревании топлива до 400—500°. Необходимо сразу же оговориться, что если справедливы теория последовательных реакций и представление о битумах как о механической «примеси» твердых минеральных топлив, то влияние процесса термического разложения топлив на механизм горения газовзвеси твердых топлив всегда будет невелико, так как после быстрого выделения летучих веществ суммарная скорость горения и газификации твердых топлив, так же как и при горении кускового топлива, будет характеризовать выгорание и газификацию частиц коксового углеродного остатка топлива, образующегося в относительно больших количествах.
В результате специальных теоретических и экспериментальных исследований автор разработал теорию термического разложения топлив, резко отличающуюся от теории последовательных реакций и дающую новое освещение результатов прежних экспериментальных исследований. В основу этой теории положено бесспорное положение, согласно которому разложение сложных «топливных молекул» в принципе не может происходить по одной реакции и не может быть даже приближенно сведено к одной реакции. В силу этого теория последовательных реакций может быть лишь частным специфическим случаем теории разложения топлива, характерным в первом приближении только для вполне определенных условий эксперимента.
Наша теория термического разложения может быть сформулирована следующим образом.
Процесс термического разложения всех топлив и их компонент протекает но ряду одновременных параллельных и последовательных реакций, имеющих во многих случаях различные кинетические характеристики. В случае термического разложения твердых топлив процесс развивается наиболее сложно и многообразно, причем так называемые «битумы» твердых «минерализованных» топлив не являются механической примесью этих топлив, а выделение их при воздействии органических растворителей на топливо есть, как правило, результат химического процесса разложения топливных молекул по наиболее слабым (в этих условиях) связям. Параллельно-последовательный ход реагирования характерен и для исходного топлива и всех промежуточных продуктов.
Эта новая теория термического разложения топлив, которую для крат
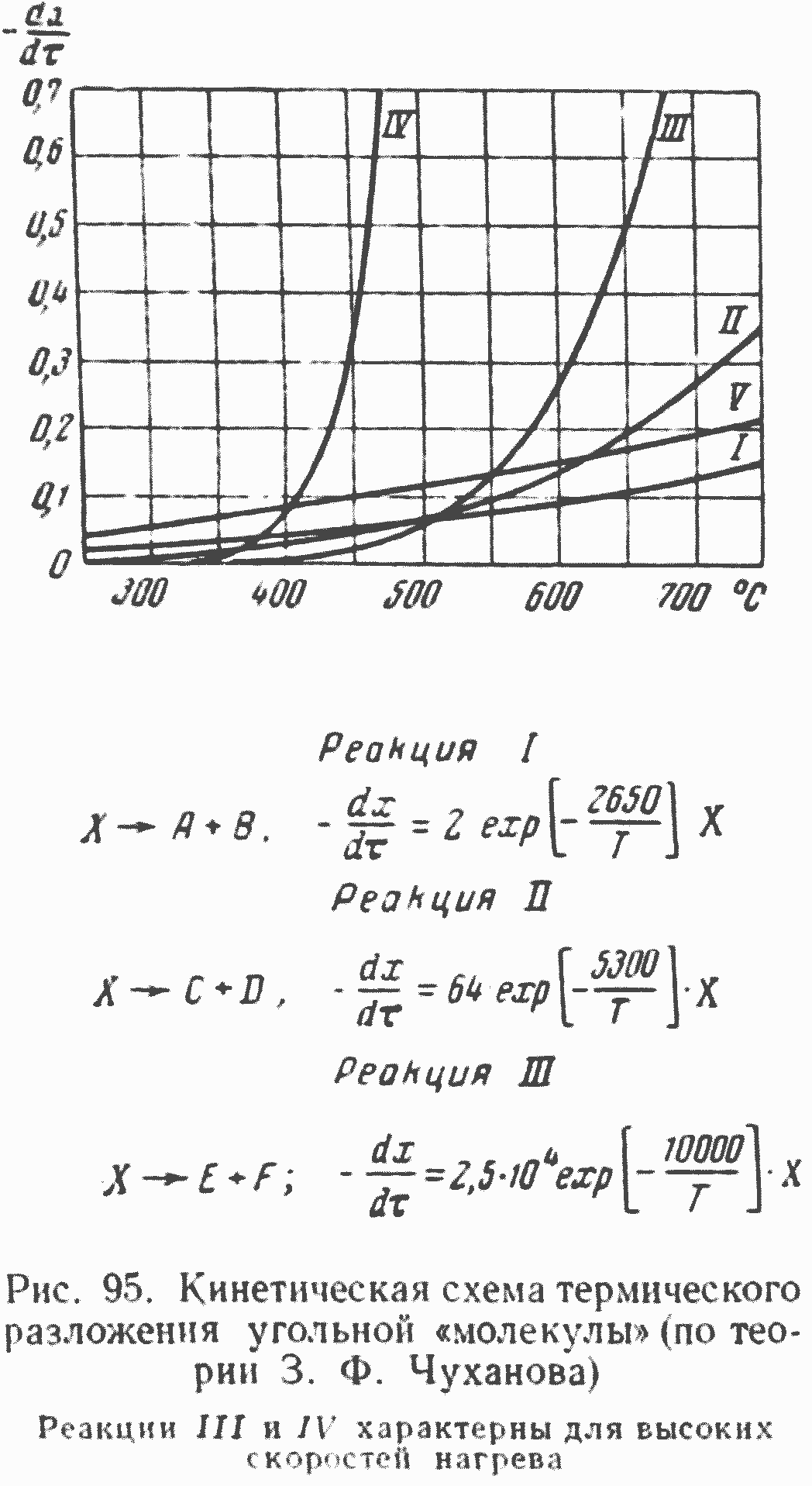
кости можно назвать «теорией параллельно-последовательных реакций», в отличие от теории Фукса—Кревелена, открывает большие возможности регулирования хода как первичных, так и вторичных реакций — разложения топливных молекул* (в т. ч. регулирования качества и выхода получаемых продуктов). Принципиальная возможность радикального вмешательства в ход процесса термического разложения топлив может быть наглядно проиллюстрирована, например, кинетическими кривыми, приведенными на рис. 95, которые показывают зависимость от температуры скорости пяти групп реакций, типичных для некоторых бурых углей. Изменения скорости этих реакций, протекающих с образованием характерных для них продуктов, обычно различны как по характеру, так и но величинам при разных температурах, что позволяет управлять выходом конкретных видов продукции, задавая соответствующие режимы реагирования, т. е. регулируя температуру и время реагирования твердых, жидких и парогазообразных веществ при заданных условиях. Для большей наглядности и более четкого представления о путях и конкретных способах регулировки процесса рассмотрим кривые изменения скоростей двух реакций, имеющих различные кинетические характеристики (рис. 96).
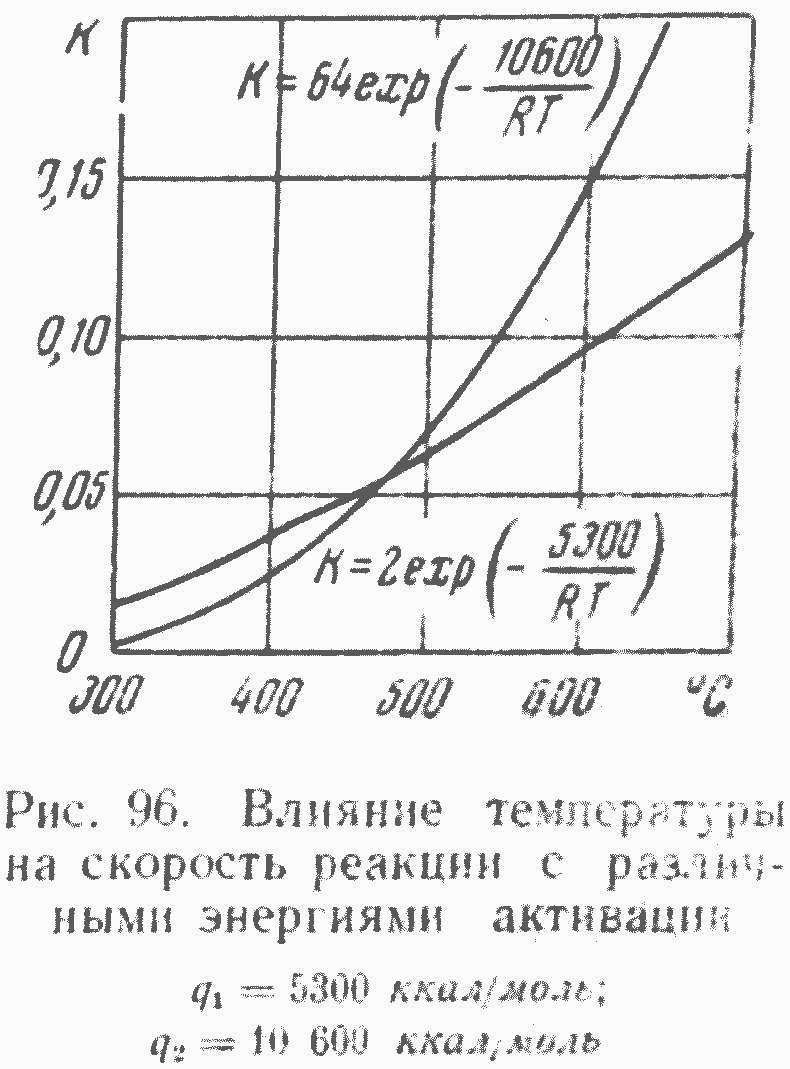
Первая реакция слабее зависит от температуры реагирования, но скорость ее практически заметна уже при низких температурах. При медленном нагреве топливного компонента, разлагающегося в случае низких температур так, как это изображено на рис. 95, все исходное вещество Л разложится по первой реакции, скорость которой в несколько раз больше второй. Следовательно, в этом случае будут получены в основном продукты первой реакции, если даже мы нагреем исходный компонент до высокой температуры. Получаемые продукты будут в этих условиях качественно отличными только за счет протекания вторичных реакций продуктов первой реакции. Это, как мы отмечали, и наблюдалось обычно при экспериментальных исследованиях с относительно медленным нагревом топливной массы.
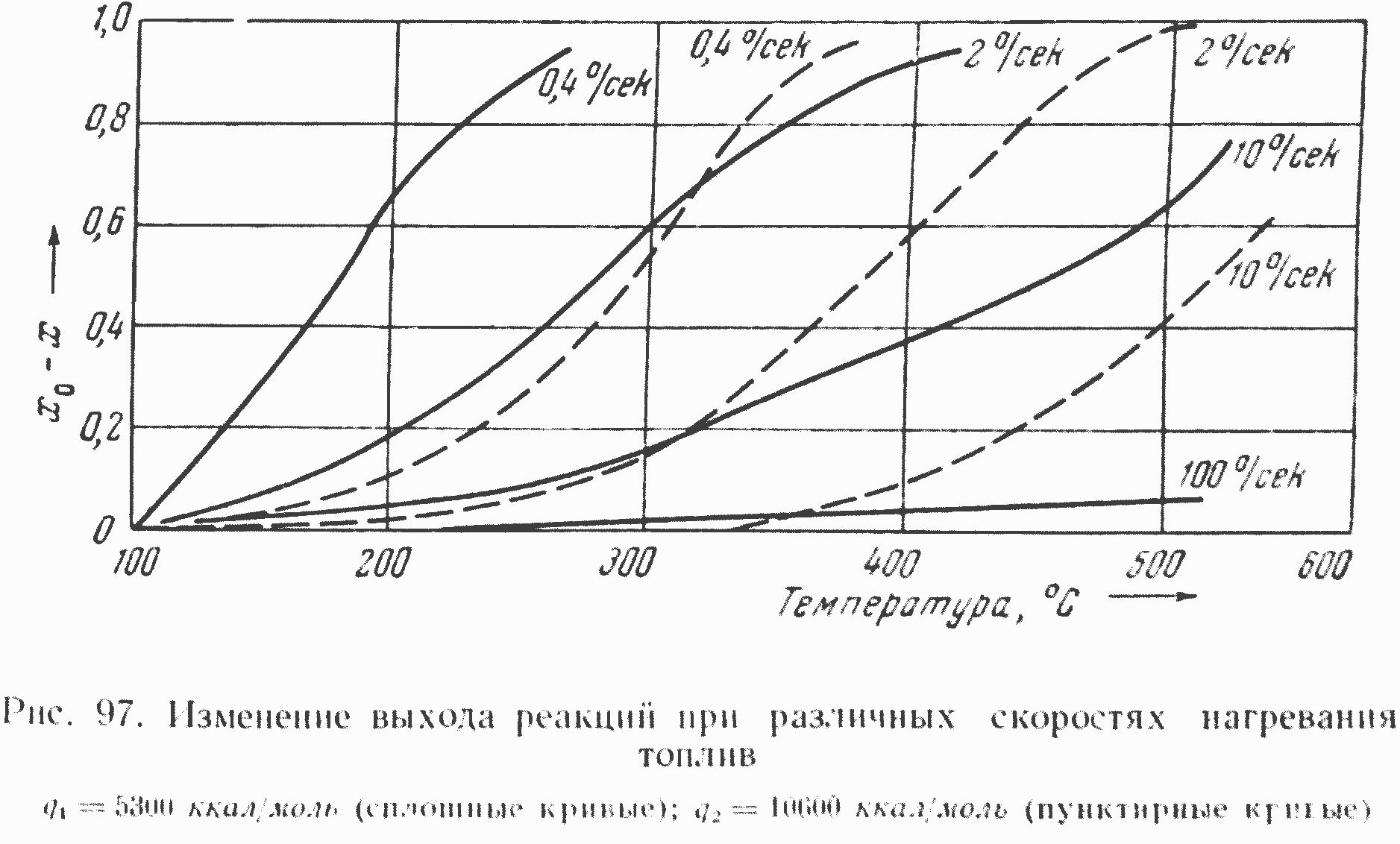
Картина развития процесса разложения при наличии двух реакций (см. рис. 96) радикально меняется, если нагрев исходного вещества до высокой температуры осуществлять с весьма большой скоростью. В этом случае исходное вещество не успевает в практически заметном количестве разложиться при низкой температуре и поэтому его разложение происходит, в основном, при той конечной температуре, до которой нагревается топливный компонент. Как видно из рис. 96, при высокой температуре скорость второй реакции значительно превышает скорость первой, и качество продуктов разложения резко изменяется: вместо продуктов первой реакции появляются продукты второй реакции. Это положение хорошо иллюстрируется рис. 97, на котором представлено развитие этих двух реакций при различных скоростях нагревания топливной компоненты и из которого видно, что при малой скорости нагрева полностью превалирует первая, а при больших скоростях — вторая реакция.
Анализ развития двух реакций при изменении температуры и скорости нагрева топлива позволяет достаточно ясно представить ход процесса и при наличии пяти и большего числа реакций. Очевидно, что теория параллельно-последовательных реакций справедлива для всех топлив, включая нефть, мазуты, смолы и природный газ, а следовательно, справедлива и для термического разложения (пиролиза) газопарообразных продуктов, получаемых из твердых топлив, т. е. для подавляющего большинства вторичных и третичных реакции разложения твердых топлив, связанных с твердыми, жидкими и газообразными продуктами термической деструкции угля, торфа, сланца, гудрона и т. д.
В том случае, когда первичное разложение конкретных «топливных молекул» (топливных компонент)* идет по одной из реакций, протекающей с высокой скоростью при сравнительно невысокой температуре, и обладающей относительно невысокой энергией активации, тогда, как правило, в практических условиях реагирование при более высоких температурах осуществляется в основном с вторичными твердыми и жидкими продуктами разложения исходного топлива.
Ниже мы обратимся к экспериментальным данным, характеризующим изменения механизма процесса и качества продуктов термического разложения при весьма высоких скоростях нагревания топливной массы, а пока кратко проанализируем теоретические возможности такого изменения и его практические результаты как в отношении продуктов термического разложения, так и в отношении изменения механизма горения и газификации пылевидного топлива.
В качестве примера возьмем органическую массу торфа (исследованного в работе Стонанса и Чуханова) с содержанием 72,8% летучих, определенных стандартным методом, и содержанием углерода, водорода и (Ο+Ν+S) соответственно 54,1; 5,6 и 40,3%. Этот торф, конечно, является смесью нескольких типов достаточно отличающихся друг от друга топливных молекул. Рассмотрим элементарный состав торфа в молярных долях; в его органической массе на 8 грамм-атомов кислорода и водорода мы имеем только 4,5 грамм-атома углерода. Таким образом, в случае образования в летучих веществах только окиси углерода, а также ароматических и непредельных углеводородов с приведенной формулой СпH2п вся органическая масса торфа превратится в летучие вещества в виде H2, СО и углеводородов. При этом на 1 кг органического вещества торфа выделится почти 200 л свободного водорода, около 50 л окиси углерода, не считая газообразных углеводородов и ароматизированных смол. При сокращении выхода свободного водорода стопроцентная «сухая» перегонка торфа1 будет происходить и при значительном выходе CO2, и даже при образовании заметных количеств пирогенетической воды, являющейся наименее «желательным» продуктом термического разложения топлив. В обычных условиях после выделения летучих веществ при термическом разложении торфа обуглероженный коксовый остаток достигает 30—45%. Практически такой результат всегда имеет место при медленном нагреве в связи с тем, что первичной торфяной смоле свойственно относительно высокое отношение Н/С, а также потому, что в газе почти не содержится непредельных углеводородов— олефинов и окиси углерода и, наоборот, превалируют метан (СН4) и CO2 и, наконец, вследствие того, что при разложении торфа в этом случае образуется достаточно большое количество первичной пирогенетической воды.
Образование всех перечисленных летучих компонентов из органического вещества торфа и приводит к минимальному переводу углерода исходного торфа в летучие вещества, что, в свою очередь, является причиной относительно высокого выхода коксового остатка. При медленном нагревании торфа уже при 500—550 С из органической массы удаляются практически весь кислород и значительная часть водорода.
1 Т. е. полное превращение торфа (без подачи дутья) в газопарообразные продукты.
Повышение выхода летучих веществ возможно, следовательно, только в том случае, если направить процесс разложения по таким реакциям, которые приводили бы к образованию летучих веществ более бедных летучеобразующими элементами — водородом и кислородом.
Если с этих позиций рассмотреть схематические изображения химических структурных формул на рис. 94, то станет очевидным, что такое направление реагирования возможно лишь при разрушении топливных молекул не путем отрыва небольших групп «шубы» топливных молекул (как это происходит при обычном медленном нагреве топлив), а путем разрыва базисной части «графитной решетки» топливных полимеров-молекул. Несомненно, что реакции разрыва молекул по базисной части связаны с более высокими энергиями активации, чем отрыв гидроксильных, карбоксильных и других групп или разрушение по кислородному «мостику». Несомненно также, что подобный процесс вообще не осуществим при медленном нагревании частиц топливной массы, так как по мере освобождения топлива от водорода и кислорода за счет очистки «шубы» топливный остаток становится все более термоустойчивым, переходя в графитизированный кокс. С другой стороны, такой разрыв связей в базисной части топливных полимеров является, очевидно, реально осуществимым только при максимальной скорости нагрева топлива до высоких температур, исключающей практически заметное реагирование топлива в период нагрева.
Определить условия и конкретный характер реакции деструкции топлива теоретически весьма затруднительно, и для этой цели необходимы специальные экспериментальные исследования; однако некоторые качественные характеристики высокоскоростного процесса термического разложения можно предопределить заранее, исходя из теоретических соображен ий.
Прежде всего теоретически можно ожидать заметного изменения качества продуктов при высокоскоростном нагреве* только при достаточно высоких температурах (более 400—550°). однако существенных и радикальных изменений процесса (получения высококачественных продуктов) следует ожидать лишь при более высоких температурах разложения топливной массы (превышающих для твердых топлив 600—650 ), когда будут осуществляться разрывы базисных связей и будет возможно образование ароматических углеводородов и олефинов. В связи с опережающим разрывом при высоких температурах базисных связей и запаздыванием отрыва боковых цепей и групп («шубы») должно иметь место повышенное содержание кислорода и водорода в твердых остатках топлива и, наконец, вполне вероятно снижение выхода и в ряде случаев запаздывание во времени образования таких кислородсодержащих соединений, как CO2 и пирогенетическая вода.
Для получения экспериментальных данных, позволяющих подтвердить и разработать теорию параллельно-последовательных реакций и доказывающих возможность управления процессом, потребовалась разработка новых специальных методов исследования, обеспечивающих быстрый и равномерный, но всей массе, высокоскоростной нагрев топлива.
Рис. 98. Влияние температуры разложения ныли подмосковного угля на состав газа, долю потенциального тепла в газе и его теплотворность при высокоскоростном нагреве и продолжительности процесса 0,35 сек.
В лаборатории, руководимой автором, такие методы и техника исследований были найдены при выполнении соответствующих работ, проведенных Николаевым, Шапатиной, Калюжным, Кашуричевым и др. Основными условиями исследования высокоскоростного процесса разложения твердых топлив являлось применение мелких частиц топлива и быстрый их нагрев излучением, твердым теплоносителем или горячим газом. Не касаясь всех исследований процесса термического разложения топлив, подтверждающих нашу теорию, остановимся только па результатах работ, наиболее ярко подтвердивших основные положения этой теории.
В работах Шапатиной, Николаева и Калюжного еще в 1949—1950 гг. было доказано, что нагрев топлива может быть осуществлен за доли секунды, причем даже при 450—500 процессы нагрева и выделения ценных летучих веществ можно разделить во времени и пространстве; замораживание топлива после нагрева позволяет сохранить в нем как жидкие продукты, так и практически всю углеводородную часть летучих веществ. Эти исследования однозначно подтвердили положение об отсутствии в углях битумов в виде самостоятельных механических компонентов твердых топлив. Это важное положение открыло перспективы регулирования процесса и создания новой высокоэффективной технологии переработки топлив.
В последующих работах Кашуричева, Стонанса и Николаева были получены наиболее наглядные результаты, подтверждающие и развивающие новую теорию термического разложения топлив.
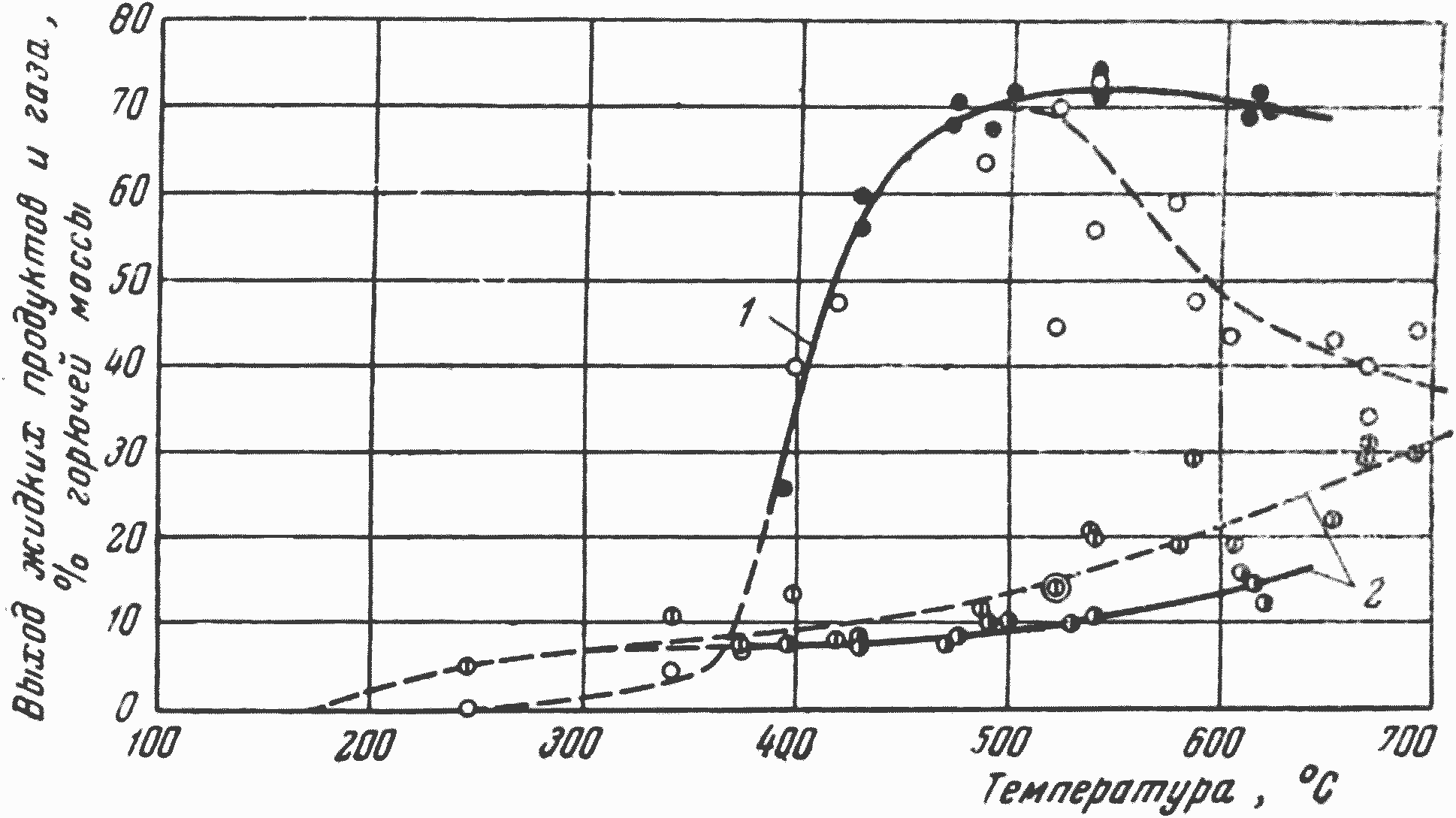
Рис. 99. Влияние температуры разложения пыли сланца (кукерсита) на выход газа и жидких продуктов (1) — дегтя, газ. бензина, пирогенетической воды и (2) — газа при изотермическом нагреве (сплошные кривые) и при наличии локальных перегревов (пунктирные кривые)
Исследуя высокоскоростной процесс термического разложения пыли бурого угля при нагреве газовым теплоносителем, Кашуричев в 1950—1951 гг. показал, как видно из рис. 98, что при повышении температуры выше 600° в получаемом газе резко (до 10%) увеличивается содержание олефиновых углеводородов. При испытании опытно-промышленной установки на бурых подмосковных углях в Калинине Николаев полностью подтвердил эти результаты, получив газ, содержавший до 15—25% этилена, пропилена и бутилена. Изучение состава газового бензина, полученного на торфе при температуре 650, показало, что в нем содержится ~ 70% бензола.
Исследования Кашуричева и Шапатиной показали, что при высокоскоростном процессе термического разложения сланцевой пыли выход смолы практически не уменьшался в случае повышения температуры до 650 (рис. 99), а при работе на торфяной пыли это уменьшение было сравнительно небольшим. Во всех исследованиях отмечено повышение содержания окиси углерода в газе при высокоскоростном термическом разложении твердых топлив.
Кашуричев, осуществляя на лабораторной установке нагрев пылевидных твердых топлив твердым теплоносителем (кварцевым песком), впервые экспериментально подтвердил возможность образования при температуре выше 600 значительных количеств пирогенетической воды после выделения смолы и основной части углеводородных газов. При этом примерно около 20—25% всей пирогенетической воды было получено в отдельном приемнике после выделения смолы; в газе этого же периода наблюдалось повышенное содержание CO2.
В работе Шапатиной, Орловой и Карасева доказано резкое снижение выхода пирогенетической воды при высокоскоростном разложении торфа.

В результате работы Стонанса. выполненной на мелкой (~10 μ) торфяной пыли, нагревавшейся за десятитысячные доли секунды (рис. 100), были получены данные о составе как газообразных, так и твердых продуктов разложения органического вещества торфа. В этой работе с наибольшей яркостью и убедительностью показано резкое различие механизма разложения торфа при медленном и высокоскоростном нагреве. Прежде всего, при высокоскоростном нагреве было выявлено образование большого количества воднорастворимых веществ, почти не возникающих при медленном нагревании. Совершенно необычным для медленного нагрева оказался состав твердых остатков, полученных при высокоскоростном процессе. Это положение наглядно иллюстрируется табл. 3, где показаны элементарный состав и выход летучих в твердых остатках торфа при разных температурах термической переработки в течение 0,1 сек.
Как видно из табл. 3, в быстро нагретом твердом остатке после выделения из него при 750'· примерно 70% летучих веществ еще сохраняется около 50% летучих. Таким образом, в опытах Стонанса наблюдалось, что почти 90% органического вещества торфа переходило в летучие вещества без. всякого реагирования торфа с воздухом или другими кислородсодержащими компонентами. Такая почти безостаточная «газификация» торфа путем его перегонки в результате высокоскоростного термического разложения является особенно интересным фактом, подтверждающим выводы теории параллельно-последовательного реагирования. Элементарный состав твердых остатков, приведенный в табл. 3, также весьма хорошо подтверждает вывод об изменении химизма разложения. При 748° в твердом «коксовом» остатке торфа еще содержится около 4% водорода и 20% кислорода. Спектрометрические исследования твердых остатков показали, что в них сохраняются гидроксильные и карбоксильные группы после выделения из торфа значительного количества смол и газообразных углеводородов, т. е. после того, как 70% органической массы торфа перешло в летучие вещества. С достаточным основанием можно предположить, что при соответствующих условиях вполне возможно практически всю органическую массу торфа перевести в летучие вещества. Этот вывод хорошо подтверждается данными табл. 4, характеризующей состав газа по опытам Стонанса при различных температурах. При 700—750 в газе содержалось около 10% олефинов (этилен, пропилен, бутилен), около 50% окиси углерода и 18—20% CO2. Таким образом, в газе присутствуют в основном компоненты, наиболее экономно расходующие главные элементы (О и Н), способные образовывать летучие вещества.
Таблица 3
Технический и элементарный анализ твердых остатков торфа при высокоскоростном нагреве (по данным Стонанса)
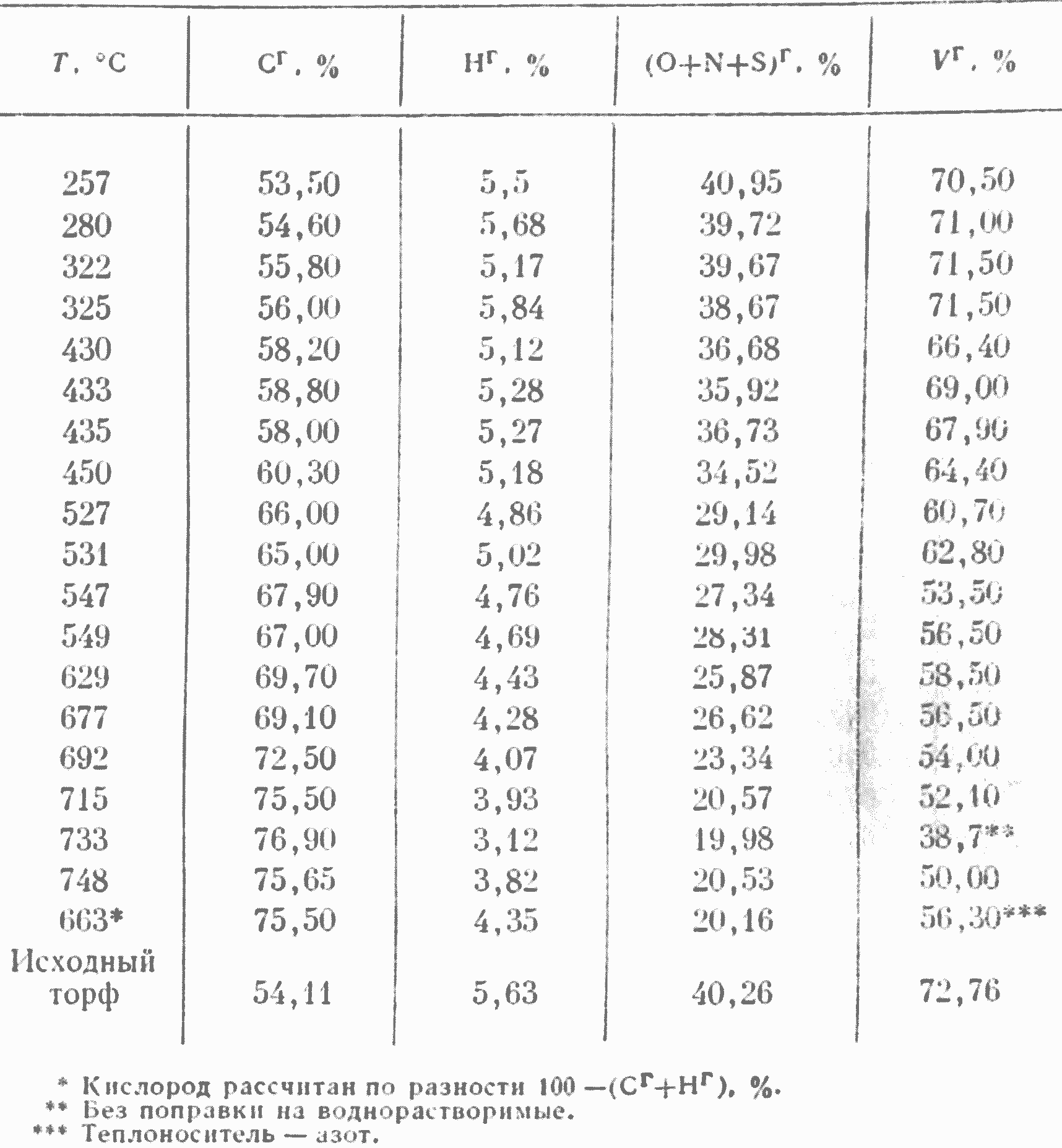
Содержание в газе метана, требующего для своего образования наибольшего количества водорода, относительно невелико и составляет 7—10% (т. е. примерно в два раза меньше, чем в газе, образующемся из торфа при медленном нагревании). Характеристики процесса, полученные на торфе, могут быть применены и для других топлив, в которых выход летучих веществ также может быть весьма сильно повышен.
Мы могли бы привести и другие, имеющиеся в настоящее время многочисленные экспериментальные данные, подтверждающие нашу теорию термического разложения топлив и одновременно убеждающие в необходимости пересмотра прежних представлений о характеристиках как процесса термического разложения, так и горения, газификации пылевидных топлив с содержанием достаточно высокого процента летучих веществ.
Насколько велико значение этого вопроса, например, для факельного сжигания газовзвеси пылевидного торфа, видно из того, что в силу исключительно быстрого нагревания пылевидных частиц торфа, поступающих в топочную камеру, возможна полная «возгонка» их — превращение в газообразное состояние до выгорания.
Таким образом, в исследуемом процессе имеет место не горение коксовых частиц (в соответствии с обычными представлениями), а горение «газообразного» топлива, образующегося из твердых частиц, так же как при сжигании жидкого топлива мы можем иметь дело только с горением паров этого топлива. В силу быстрого подъема температуры твердых частиц как за счет излучения, так и за счет подсоса горячих газов в факеле, из горящих частиц должны образовываться самые разнообразные продукты, в том числе и весьма ценные. В силу этих же причин такой характер горения
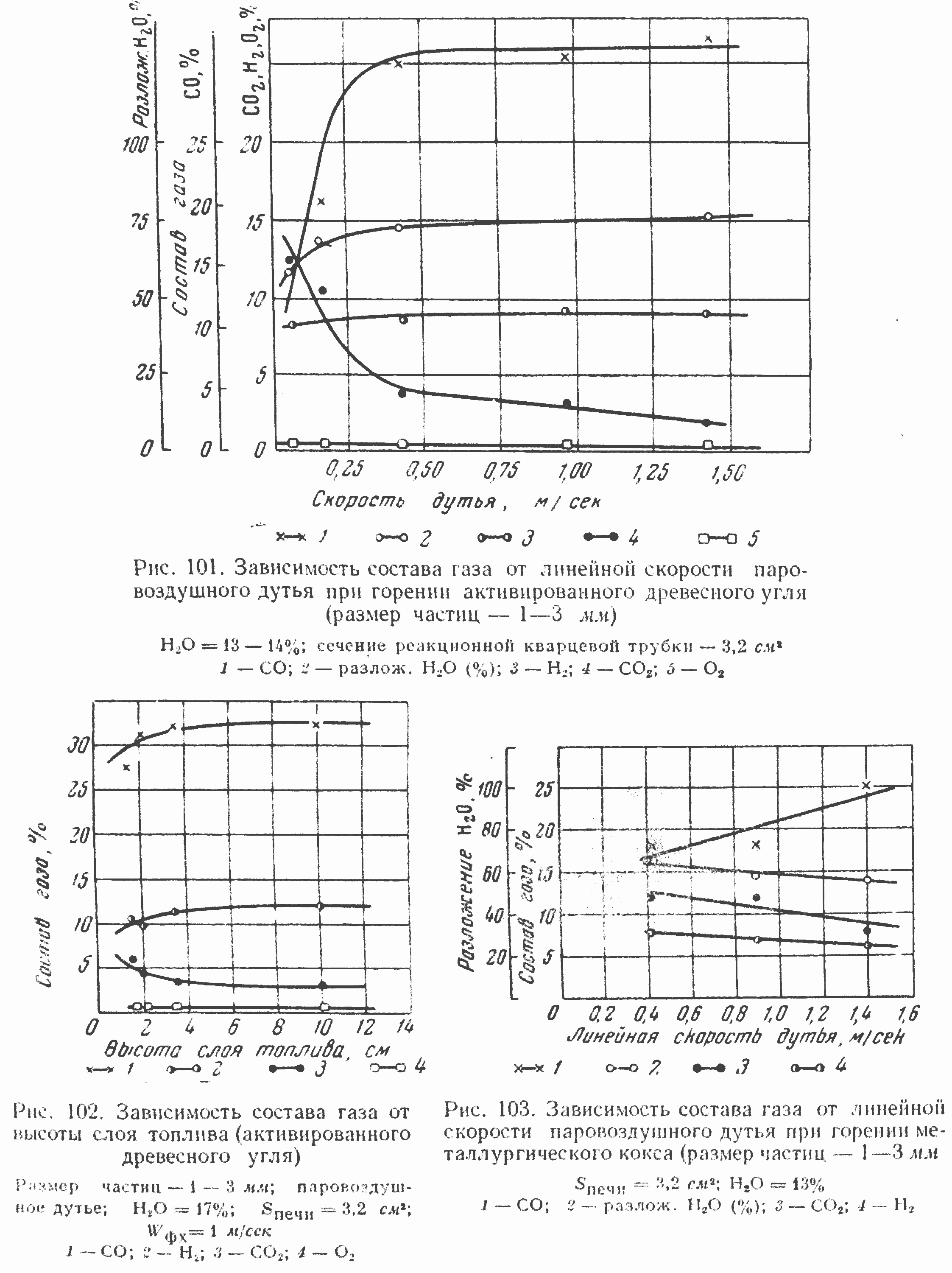
Особенно ярко отличие в процессе разложения H2О и CO2 в кислородной зоне видно на рис. 103, где показано на примере горения кокса изменение состава газа при увеличении скорости паровоздушного дутья. Сравнивая результаты, полученные при горении кокса с применением дутья «воздух + водяной пар», с результатами, представленными на рис. 70 (дутье «воздух + CO2») (стр. 116), можно убедиться, что при одинаковых условиях H2О разлагается на 50%, а CO2 практически не разлагается.
Характерным для процесса высокоскоростного горения (газификации) кокса на паровоздушном дутье является то, что степень разложения пара снижается относительно медленно при изменении концентрации H2О от 5 до 27% (рис. 104). При применении парокислородного дутья повышение концентрации пара до 50% позволяет на древесном угле получить при высокоскоростной газификации газ с 30% H2 и 60% СО (практически полное разложениe водяного пара).
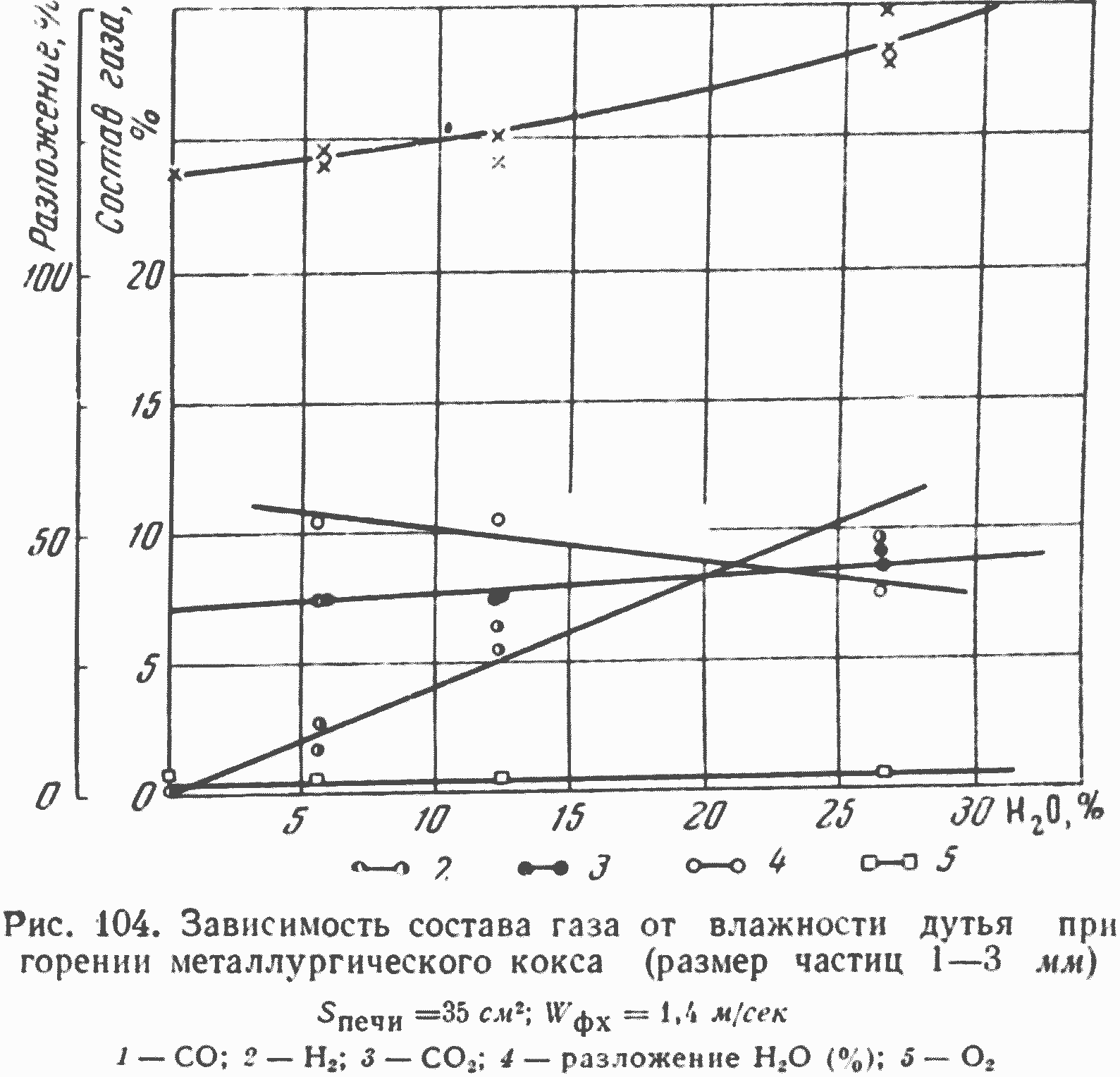
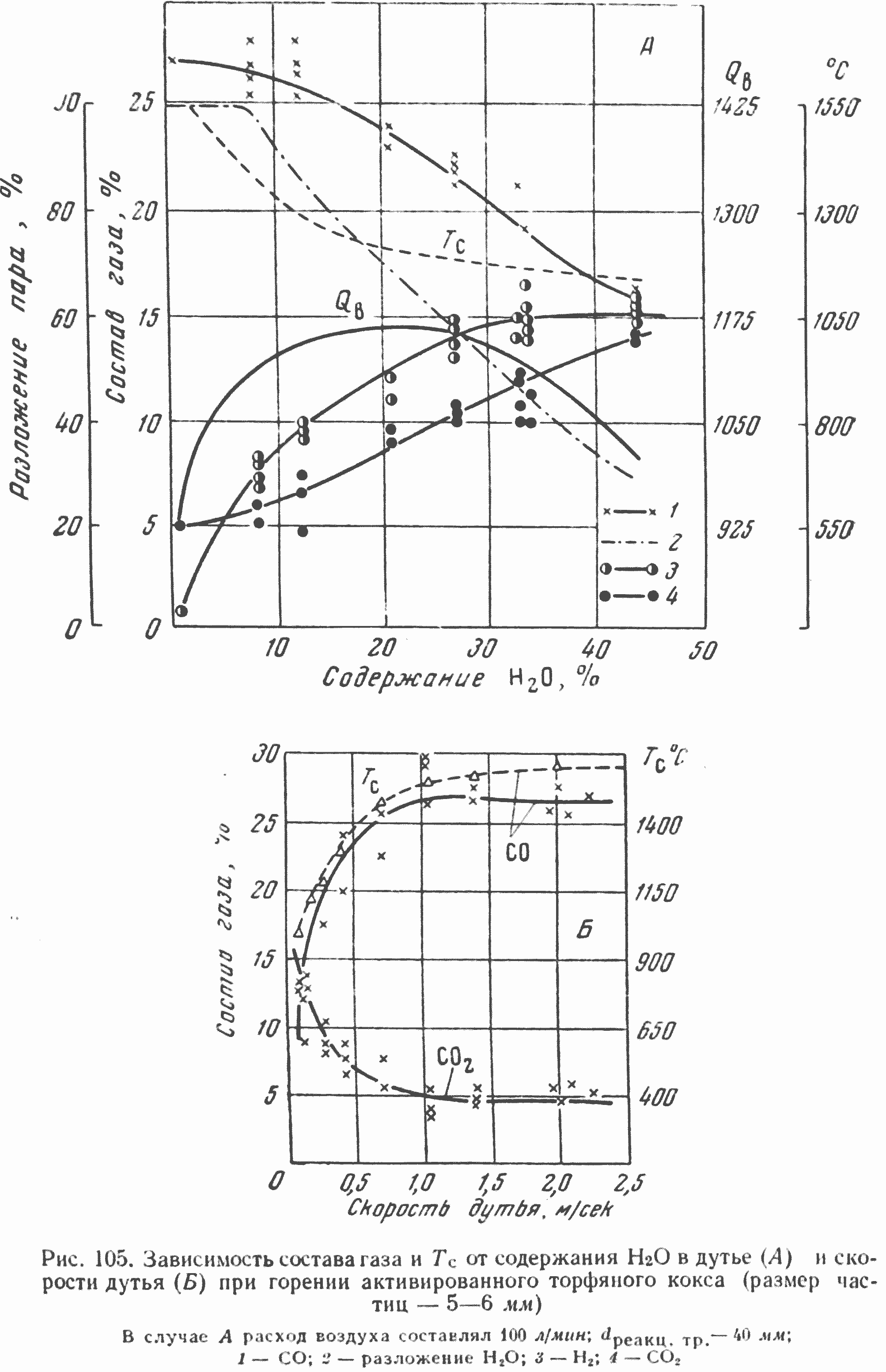
Исследования, выполненные Лавровым [11] по высокоскоростной газификации торфяного кокса на паро-воздушном дутье с замерами температур, подтвердили, что высокая интенсивность разложения пара в кислородной зоне наблюдается при сравнительно невысоких температурах, порядка 1150—1250° (рис. 105, А). Если сопоставить скорости разложения водяного пара в кислородной и восстановительной зонах, то будет очевидно, что мы имеем дело здесь не с одной и той же реакцией, а с двумя, совершенно различными. На рис. 105, Б показано влияние скорости дутья на газообразование при высокоскоростной газификации торфяного кокса на воздушном дутье.
Мы не будем здесь подробно останавливаться на вероятном механизме этих реакций, отметим только, что по нашей схеме 120] реакция разложения водяного пара в кислородной зоне, названная нами реакцией пароокисления, протекает, аналогично окислению углерода, следующим образом:
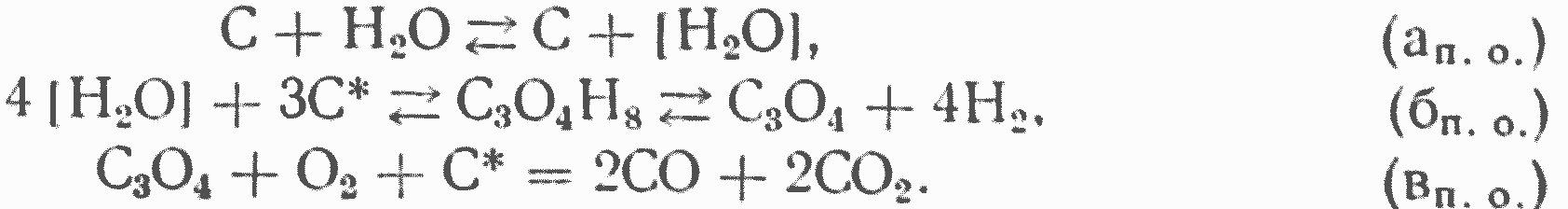
В связи с высокой скоростью адсорбции водяного пара углеродом* скорость реакции пароокисления должна быть достаточно высокой и даже сравнимой со скоростью взаимодействия углерода с кислородом по реакции окисления, которая, как мы отмечали, сама протекает только в присутствии водяных паров.
*Особенно активными формами углерода типа активированного древесного угля.
В случае, если реакция пароокисления протекает в соответствии с представленной схемой, то на каждые четыре молекулы образующегося в продуктах реакции водорода должна расходоваться в кислородной зоне одна молекула кислорода и получаться две молекулы окиси и две молекулы двуокиси углерода.
В работе Чуханова и Каржавиной были соответствующим образом обработаны экспериментальные данные по газообразованию при горении слоя древесного угля на паровоздушном дутье.
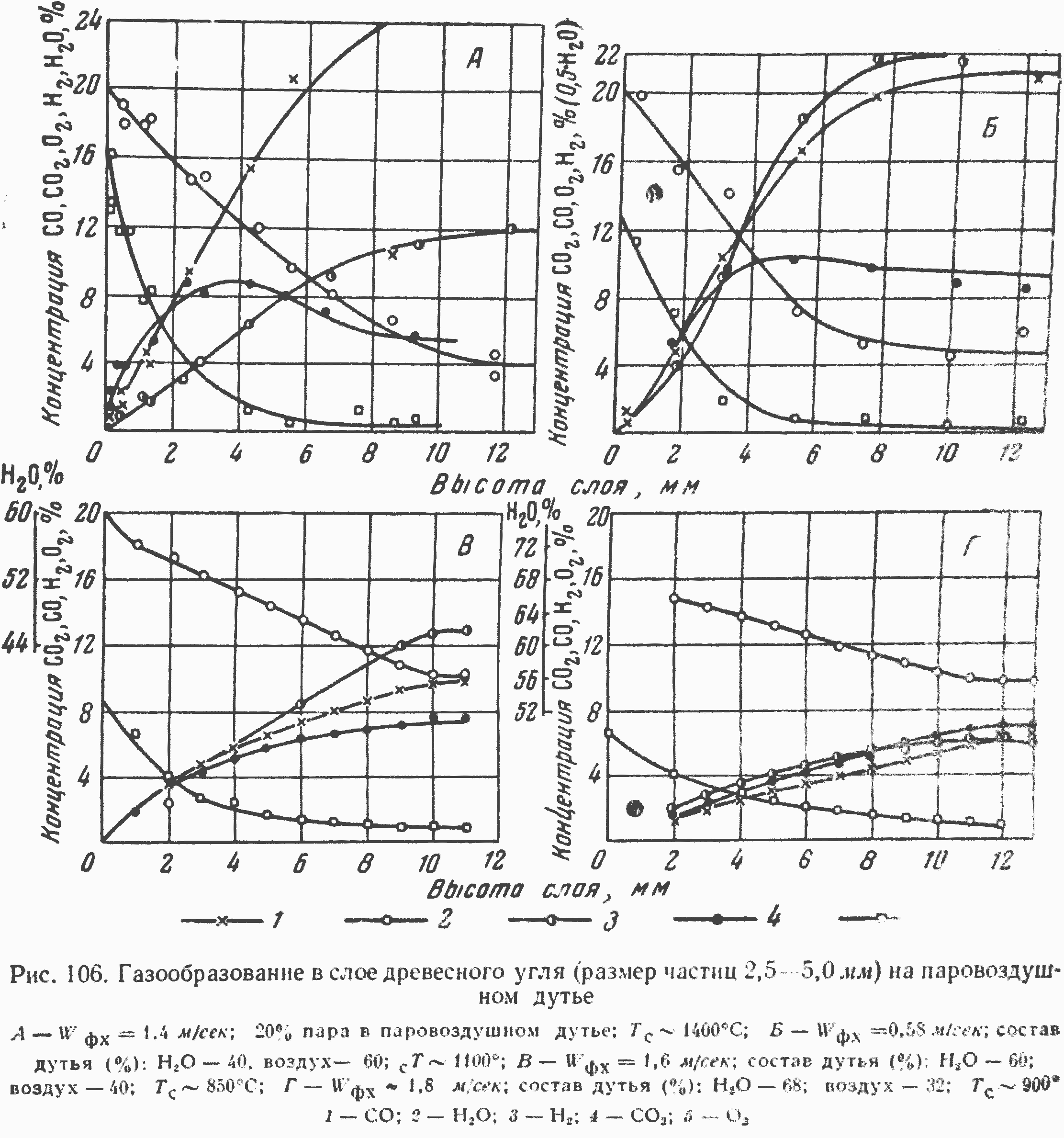
На рис. 106 приведены экспериментальные данные, полученные Каржавиной при различных концентрациях водяного пара в дутье и различных температурах реагирования. Кривые, приведенные на рисунках, убедительно подтверждают, что разложение водяного пара при высокоскоростной газификации происходит в пределах кислородной зоны, и образующийся при этом водород при высоких скоростях дутья выносится из кислородной зоны вместе с окисью углерода. Таким образом, на паровоздушном дутье, несмотря на снижение концентрации кислорода, выгорание углерода в кислородной зоне частично происходит за счет реакции пароокисления. Однако это явление наблюдается только при высокоскоростном процессе, обеспечивающем вынос несгоревших СО и H2 из кислородной зоны. В случае, если СО и H2 сгорают в объеме газа в кислородной зоне, длина ее сокращается, если же горение СО и H2 осуществляется в непосредственной близости к поверхности углерода, размер кислородной зоны и интенсивность выгорания углерода остаются неизменными, как и на сухом дутье с пониженной концентрацией кислорода.
Представленные на рисунках данные о газообразовании в слое древесного угля на паровоздушном дутье иллюстрируют динамику расходования кислорода по слою горящего угля и дают возможность приближенно рассчитывать соотношение окислов углерода, образующихся при пароокислении. Такой расчет показывает, что процесс разложения H2О в кислородной зоне сопровождается эквивалентным выходом обоих окислов углерода.
Как видно из рис. 106. А, Б, при низких концентрациях водяного пара в дутье степень разложения пара высока. При повышении концентрации паров в воздухе степень их разложения начинает заметно снижаться в связи с недостатком кислорода, потребляемого при реакциях горения, окисления и пароокисления углерода. Распределение кислорода между указанными реакциями идет в соответствии с их кинетическими характеристиками. Следует отметить, что уже при концентрации водяного пара в воздухе около 45%, при полном разложении пара в кислородной зоне, реакция пароокисления поглощала бы весь кислород, что, конечно, невозможно, так как кислород расходуется и на реакции окисления и горения углерода. Повышение концентрации кислорода в дутье позволяет обеспечить 100-процентное разложение пара при весьма высоких начальных концентрациях его. Снижение температуры реагирования и концентрации кислорода при добавке пара в воздушное дутье приводит к снижению роли реакции горения углерода и, как видно из рис. 106, Г, при концентрации водяного пара — 68% и температуре 900 соотношение окислов углерода близко к единице, что свидетельствует о практически полном прекращении реакции горения углерода. Дальнейшее исследование реакции пароокисления, несомненно, позволит установить кинетические характеристики ее и связь с реакцией окисления углерода.
Естественно, возникает вопрос о том, в какой области гетерогенного реагирования протекает реакция пароокисления в условиях высокоскоростной газификации. Ответить на него точно можно будет только после получения кинетических характеристик этой реакции, однако можно предполагать с достаточной достоверностью, что реакция пароокисления на активированном древесном угле, как и реакция окисления мелких частиц углерода, находится в основном в переходной области гетерогенного реагирования.
Процесс высокоскоростной газификации на паровоздушном дутье, как и на воздушном, осуществим, как мы указывали, только в том случае, если горение СО и H2 идет по первому (I) кинетическому уравнению (стр. 93) (невоспламененное взаимодействие с кислородом), т. е. в слое с мелкими каналами между частицами. Для сферических частиц критический размер куска составляет 7—10 мм, для частиц неправильной формы и особенно смеси кусков различного размера этот максимальный предельный размер в случае осуществления высокоскоростной газификации будет большим.
*См. раздел 1 этой главы.